Page d'accueil
Résumé de la Thèse ayant obtenu le PRIX GREMI 2010 :
Signalisation de TLR2, organisation et variabilité de la réponse immunitaire innée
Julie TOUBIANA
Ce travail a été réalisé à l' Institut Cochin INSERM U1016 / CNRS UMR 8104

Cette approche nous a permis d’identifier et de caractériser le rôle de la tyrosine kinase Lyn et de l’enzyme IMPDHII (Inosine 5’ Monophosphate Dehydrogenase II) dans la signalisation de TLR2 (Figure 2 & 3). Lyn participe à l’activation de NF-κB après engagement de TLR2 via l’activation de la PI3-Kinase et la transactivation de la sous-unité p65 de NF-κB (Figure 2). I MPDHII participe à la régulation négative de TLR2 à travers l’augmentation de l’activité phosphatase de SHP1 et la déphosphorylation de la sous-unité p85 de la PI3-Kinase (Figure 3). Le mécanisme exact par lequel IMPDHII régule l’activité de SHP1 reste à élucider (3).
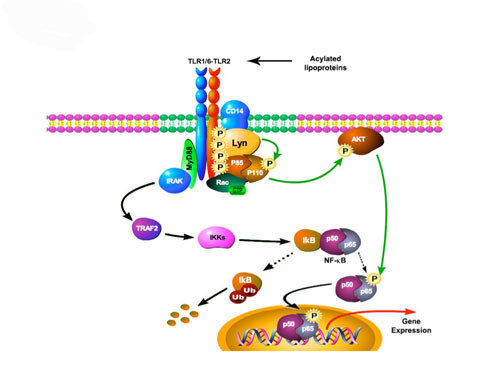
Figure 2. Lyn contrôle la voie d’activation de la PI3-K par la phosphorylation de sa sous-unité p110 engagement de TLR2 . La présence de lipopeptides bactériens entraîne l’hetero-dimérisation de TLR1/6-TLR2 au sein des microdomaines membranaires. MyD88/IRAK sont recrutés au récepteur et permettent la dégradation d’I-κB et la translocation nucléaire de NF-κB. Une voie dépendante de Rac1 et PI3-K implique Lyn et CD14 au sein d’un cluster membranaire d’activation. Après phosphorylation de TLR2, la sous-unité p85 de PI3-K est recrutée au récepteur et phosphorylée sur tyrosine. Une phosphorylation dépendante de Lyn sur tyrosine de sa sous-unité catalytique p110 permet l’activation de la PI3-K afin de recruter AKT et d’aboutir à la transactivation de la sous-unité p65 de NF-κB et à l’expression des gènes de l’inflammation.
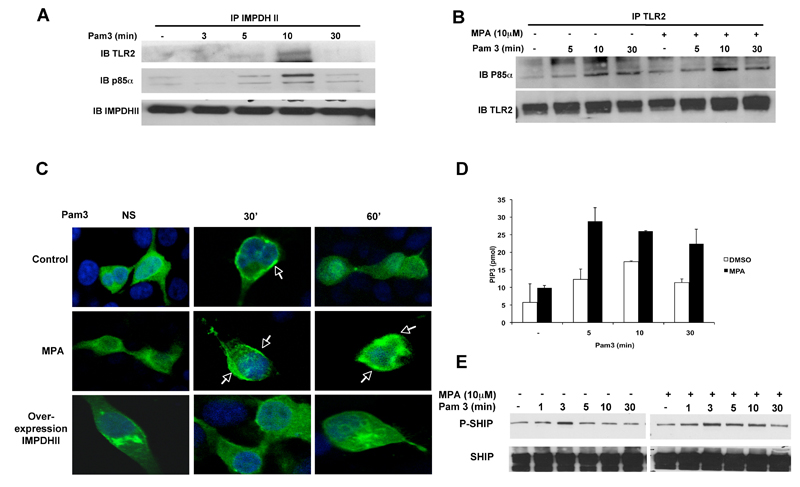
Figure 3. IMPDHII régule négativement l’activité de la PI3-Kinase. Afin de déterminer le rôle de IMPDHII dans l’activité de PI-3K, le recrutement de Akt vers le phospholipide membranaire PIP3 a été étudié par microscopie confocale. Les cellules HEK 293-TLR2 ont été transfectées avec une construction codant pour une protéine de fusion entre le domaine PH d’Akt et le marqueur fluorescent GFP. Dans les cellules contrôles, la stimulation de TLR2 par Pam3 induit une translocation transitoire de la sonde fluorescente à la membrane plasmique entre 30 et 45 min après stimulation. L’inhibition de l’activité de IMPDHII avec le MPA entraîne un recrutement persistant d’Akt sur le PIP3, alors que la surexpression de IMPDHII empêche le recrutement d’Akt à la membrane plasmique, suggérant que IMPDHII régule négativement l’activité de la PI3-Kinase.
Enfin, par une approche transrationnelle, nous avons étudié le rôle d’un polymorphisme du gène IRAK1 , dont la protéine est essentielle à la signalisation de la majorité des TLRs, dans la variabilité du phénotype clinique chez des patients septiques. Ce travail a permis de mettre en évidence que l’haplotype fonctionnel d’IRAK1 (avec gain de fonction), représenté par le SNP-Tag IRAK1- 1595C , est associé à la sévérité de l’atteinte pulmonaire au cours du choc septique dans une cohorte de plus de 800 patients (4). Ce travail introduit des perspectives nouvelles sur l’organisation, la régulation et la variabilité de la réponse immunitaire innée. Il souligne l’importance de l’équilibre nécessaire entre signaux activateurs et inhibiteurs coordonnant la réponse inflammatoire, et pourrait à plus long terme nous aider à identifier de nouvelles cibles thérapeutiques au cours du sepsis.
Références :
1. Kawai, T., and Akira, S. (2010) Nat Immunol 11 , 373-384
2. Triantafilou, M., Gamper, F. G., Haston, R. M., Mouratis, M. A., Morath, S., Hartung, T., and Triantafilou, K. (2006) J Biol Chem 281 , 31002-31011
3. Toubiana, J., Rossi, A. L., Grimaldi, D., Belaidouni, N., Chafey, P., Clary, G., Courtine, E., Pene, F., Mira, J. P., Claessens, Y. E., and Chiche, J. D. (2011) J Biol Chem
4. Toubiana, J., Courtine, E., Pene, F., Viallon, V., Asfar, P., Daubin, C., Rousseau, C., Chenot, C., Ouaaz, F., Grimaldi, D., Cariou, A., Chiche, J. D., and Mira, J. P. (2010) Crit Care Med 38 , 2287-2294